python 服务器批处理得到PSSM矩阵的问题
目录
- 1. 在linux上安装psiblast
- 2.下载并编译用于比对的大型蛋白质数据库
- 3. 获取PSSM矩阵
- 1)单条蛋白质序列的处理方法
- 2)批处理获取的方法
- 参考文献:
1. 在linux上安装psiblast
最好新建一个python环境,因为我发现conda安装blast默认的是python==3.6.11,可能会不小心把你的python版本改掉…然后你写好的代码全die了……
conda create -n blast python==3.6.11 source activate blast conda install -c bioconda blast
2.下载并编译用于比对的大型蛋白质数据库
nr和uniprot是比较通用的数据库:
ftp://ftp.ncbi.nlm.nih.gov/blast/db/
https://www.uniprot.org/downloads
1)nr是ncbi收集的目前所有微生物的蛋白序列,是用来计算氨基酸一般情况下的频率的,160G
2)uniprot90根据相似性做了一个去冗余,所以比nr要小很多,56G
# 以uniprot90为例 wget ftp://ftp.uniprot.org/pub/databases/uniprot/uniref/uniref90/uniref90.fasta.gz # 下载 gzip -d uniref90.fasta.gz # 解压 makeblastdb -in uniref90.fasta -parse_seqids -hash_index -dbtype prot # 编译
解析完成后的样子:
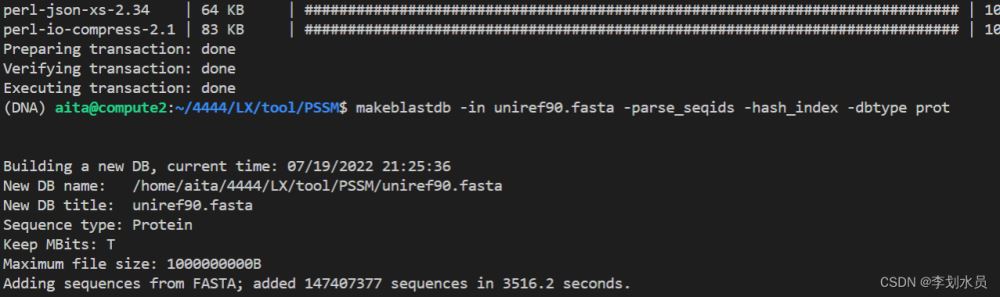
文件是这个样子:(只截取了一部分)
3. 获取PSSM矩阵
我的初始文件是:
P00269.fasta是对单条蛋白质处理,里面的格式是:

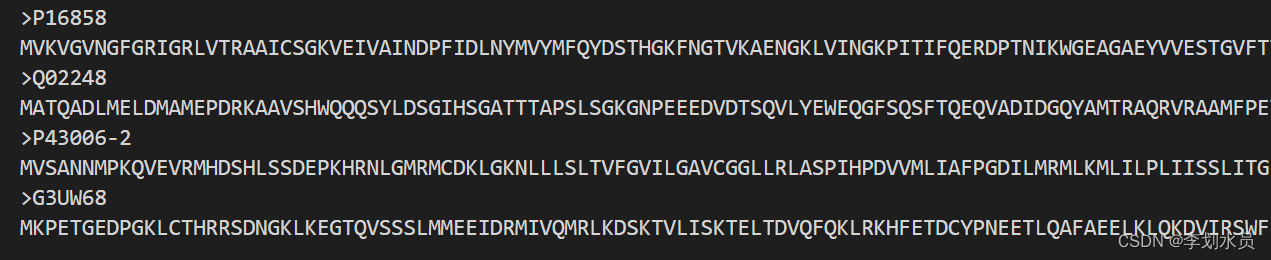
testset.fasta是对蛋白质集合批处理,里面的格式是(也可以单独蛋白质存为.fasta文件,由于blast只能处理单条蛋白糊,把这个集合知识归总的意思,第一步还是要生成单条蛋白质的.fasta文件,所以这个文件看个人意愿):
1)单条蛋白质序列的处理方法
import os
os.system('psiblast -query dataset/P00269.fasta -db /PSSM/uniref90.fasta -num_iterations 3 -out_ascii_pssm /dataset/P00269.pssm')##这个蛋白质好慢呀
2)批处理获取的方法
import os
file_name='/dataset/testset.fasta'
Protein_id=[]
with open(file_name,'r') as fp:
i=0
for line in fp:
if i%2==0:
# Protein_id.append(line[1:-1])
id=line[0:-1]
p=line[1:-1]
with open ('/dataset/'+str(p)+'.fasta','a') as protein:
protein.write(id)
# protein.write()
if i%2==1:
seq=line[0:-1]
with open ('/dataset/'+str(p)+'.fasta','a') as protein:
protein.write('\n')
protein.write(seq)
i=i+1
os.system('psiblast -query '+'/dataset/'+str(p)+'.fasta -db /PSSM/uniref90.fasta -num_iterations 3 -out_ascii_pssm /dataset/'+str(p)+'.pssm')
##PSSM真是太慢了,下面是只生成一个后的截图
emmmm,在研究怎么把这个矩阵存入文件方便调用,今天应该会更新……但是他好慢啊,不想用了。
参考文献:
到此这篇关于python服务器批处理得到PSSM矩阵的文章就介绍到这了,更多相关python服务器批处理内容请搜索我们以前的文章或继续浏览下面的相关文章希望大家以后多多支持我们!
相关推荐
-

Python实现两种多分类混淆矩阵
目录 1.什么是混淆矩阵 2.分类模型评价指标 3.两种多分类混淆矩阵 3.1直接打印出每一个类别的分类准确率. 3.2打印具体的分类结果的数值 4.总结 1.什么是混淆矩阵 深度学习中,混淆矩阵是ROC曲线绘制的基础,同时它也是衡量分类型模型准确度中最基本,最直观,计算最简单的方法.它可以直观地了解分类模型在每一类样本里面表现,常作为模型评估的一部分.它可以非常容易的表明多个类别是否有混淆(也就是一个class被预测成另一个class). 首先要明确几个概念: T或者F:该样本 是否被正确分类
-
python 共现矩阵的实现代码
目录 python共现矩阵实现 项目背景 什么是共现矩阵 共现矩阵的构建思路 共现矩阵的代码实现 共现矩阵(共词矩阵)计算 共现矩阵(共词矩阵) 补充一点 python共现矩阵实现 最近在学习python词库的可视化,其中有一个依据共现矩阵制作的可视化,感觉十分炫酷,便以此复刻. 项目背景 本人利用爬虫获取各大博客网站的文章,在进行jieba分词,得到每篇文章的关键词,对这些关键词进行共现矩阵的可视化. 什么是共现矩阵 比如我们有两句话: ls = ['我永远喜欢三上悠亚', '三上悠亚又出新作
-
Python中的Numpy 矩阵运算
目录 在学习线性代数时我们所接触的矩阵之间的乘法是矩阵的叉乘,有这样一个前提: 若矩阵A是m*n阶的,B是p*q阶的矩阵,AB能相乘,首先得满足:n=p,即A的列数要等于B的行数.运算的方法如下图: 当时学线性代数时老师教的更为直观记法: 点乘则是这样: 假如有a,b两个矩阵,在Matlab中我们实现点乘和叉乘的方式分别如下: a.*b %表示点乘 a*b %表示叉乘 下面我们来看看python中的操作: import numpy as np a = np.arange(1, 10).resha
-
python 服务器批处理得到PSSM矩阵的问题
目录 1. 在linux上安装psiblast 2.下载并编译用于比对的大型蛋白质数据库 3. 获取PSSM矩阵 1)单条蛋白质序列的处理方法 2)批处理获取的方法 参考文献: 1. 在linux上安装psiblast 最好新建一个python环境,因为我发现conda安装blast默认的是python==3.6.11,可能会不小心把你的python版本改掉…然后你写好的代码全die了…… conda create -n blast python==3.6.11 source activate
-
python服务器与android客户端socket通信实例
本文实例讲述了python服务器与android客户端socket通信的方法.分享给大家供大家参考.具体实现方法如下: 首先,服务器端使用python完成,下面为python代码: 复制代码 代码如下: #server.py import socket def getipaddrs(hostname):#只是为了显示IP,仅仅测试一下 result = socket.getaddrinfo(hostname, None, 0, socket.SOCK_STREAM) re
-
Python numpy生成矩阵、串联矩阵代码分享
import numpy 生成numpy矩阵的几个相关函数: numpy.array() numpy.zeros() numpy.ones() numpy.eye() 串联生成numpy矩阵的几个相关函数: numpy.array() numpy.row_stack() numpy.column_stack() numpy.reshape() >>> import numpy >>> numpy.eye(3) array([[ 1., 0., 0.], [ 0., 1.
-
Python的numpy库中将矩阵转换为列表等函数的方法
这篇文章主要介绍Python的numpy库中的一些函数,做备份,以便查找. (1)将矩阵转换为列表的函数:numpy.matrix.tolist() 返回list列表 Examples >>> >>> x = np.matrix(np.arange(12).reshape((3,4))); x matrix([[ 0, 1, 2, 3], [ 4, 5, 6, 7], [ 8, 9, 10, 11]]) >>> x.tolist() [[0, 1, 2
-
Python 读取图片文件为矩阵和保存矩阵为图片的方法
读取图片为矩阵 import matplotlib im = matplotlib.image.imread('0_0.jpg') 保存矩阵为图片 import numpy as np import scipy x = np.random.random((600,800,3)) scipy.misc.imsave('meelo.jpg', x) 以上这篇Python 读取图片文件为矩阵和保存矩阵为图片的方法就是小编分享给大家的全部内容了,希望能给大家一个参考,也希望大家多多支持我们. 您可能感兴
-
python 读取文件并把矩阵转成numpy的两种方法
在当前目录下: 方法1: file = open('filename') a =file.read() b =a.split('\n')#使用换行 len(b) #统计有多少行 for i in range(len(b)): b[i] = b[i].split()#使用空格分开 len(b[0])#可以查看第一行有多少列. B[0][311]#可以查看具体某行某列的数 import numpy as np b = np.array(b)#转成numpy形的 type(b) # 输出<输出clas
-
Python解决线性代数问题之矩阵的初等变换方法
定义一个矩阵初等行变换的类 class rowTransformation(): array = ([[],[]]) def __init__(self,array): self.array = array def __mul__(self, other): pass # 交换矩阵的两行 def exchange_two_lines(self,x,y): a = self.array[x-1:x].copy() self.array[x-1:x] = self.array[y-1:y] self
-
编写多线程Python服务器 最适合基础
编写一个多线程的Python服务器. 多线程Python服务器使用以下主要模块来管理多个客户端连接. 1. Python的线程模块 2. SocketServer的 ThreadingMixIn 上述两个模块中的第二个类使得Python服务器能够分叉新线程来照顾每一个新的连接.它也使程序异步运行线程. 这个多线程Python服务器程序包括以下三个Python模块. 1. Python-Server.py 2. Python-ClientA.py 3. Python-ClientB.py Pyth
-
Python使用numpy模块实现矩阵和列表的连接操作方法
Numpy模块被广泛用于科学和数值计算,自然有它的强大之处,之前对于特征处理中需要进行数据列表或者矩阵拼接的时候都是自己写的函数来完成的,今天发现一个好玩的函数,不仅好玩,关键性能强大,那就是Numpy模块自带的矩阵.列表连接函数,实践一下. #!usr/bin/env python #encoding:utf-8 from __future__ import division ''' __Author__:沂水寒城 使用numpy模块实现矩阵的连接操作 ''' import numpy as
-
在python Numpy中求向量和矩阵的范数实例
np.linalg.norm(求范数):linalg=linear(线性)+algebra(代数),norm则表示范数. 函数参数 x_norm=np.linalg.norm(x, ord=None, axis=None, keepdims=False) ①x: 表示矩阵(也可以是一维) ②ord:范数类型 向量的范数: 矩阵的范数: ord=1:列和的最大值 ord=2:|λE-ATA|=0,求特征值,然后求最大特征值得算术平方根 ord=∞:行和的最大值 ③axis:处理类型 axis=1表