使用Python脚本提取基因组指定位置序列
引言
在基因组分析中,我们经常会有这么一个需求,就是在一个fasta文件中提取一些序列出来。有时这些序列是一段完整的序列,而有时仅仅为原fasta文件中某段序列的一部分。特别是当数据量很多时,使用肉眼去挑选序列会很吃力,那么这时我们就可以通过简单的编程去实现了。
例如此处在网盘附件中给定了某物种的全基因组序列(0-refer/ Bacillus_subtilis.str168.fasta),及其基因组gff注释文件(0-refer/ Bacillus_subtilis.str168.gff)。
假设在这里我们对该物种进行研究,通过gff注释文件中的基因功能描述字段,加上对相关资料的查阅等,定位到了一些特定的基因。
接下来我们期望基于gff文件中对这些基因位置的描述,在全基因组序列fasta文件中将这些基因找到并提取出来,得到一个新的fasta文件,新文件中只包含目的基因序列。
请使用python3编写一个可以实现该功能的脚本。
示例
一个示例脚本如下(可参见网盘附件“seq_select1.py”)。
为了实现以上目的,我们首先需要准备一个txt文件(以下称其为list文件,示例list.txt可参见网盘附件),基于gff文件中所记录的基因位置信息,填入类似以下的内容(列与列之间以tab分隔)。
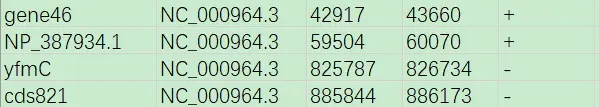
#下列内容保存到list.txt gene46 NC_000964.3 42917 43660 + NP_387934.1 NC_000964.3 59504 60070 + yfmC NC_000964.3 825787 826734 - cds821 NC_000964.3 885844 886173 -
第1列,给所要获取的新序列命个名称;
第2列,所要获取的序列所在原序列ID;
第3列,所要获取的序列在原序列中的起始位置;
第4列,所要获取的序列在原序列中的终止位置;
第5列,所要获取的序列位于原序列的正链(+)或负链(-)。
之后根据输入文件,即输入fasta文件及记录所要获取序列位置的list文件中的内容,编辑py脚本。
打开fasta文件“Bacillus_subtilis.scaffolds.fasta”,使用循环逐行读取其中的序列id及碱基序列,并将每条序列的所有碱基合并为一个字符串;将序列id及该序列合并后的碱基序列以字典的形式存储(字典样式{'id':'base'})。
打开list文件“list.txt”,读取其中的内容,存储到字典中。字典的键为list文件中的第1列内容;字典的值为list文件中第2-5列的内容,并按tab分割得到一个列表,包含4个字符分别代表list文件中第2-5列的信息)。
最后根据读取的list文件中序列位置信息,在读取的基因组中截取目的基因序列。由于某些基因序列可能位于基因组负链中,需取其反向互补序列,故首先定义一个函数rev(),用于在后续调用得到反向互补序列。在输出序列名称时,还可选是否将该序列的位置信息一并输出(name_detail = True/False)。
<pre class="r" style="overflow-wrap: break-word; font-style: normal; font-variant-ligatures: normal; font-variant-caps: normal; font-weight: 400; letter-spacing: normal; orphans: 2; text-align: start; text-indent: 0px; text-transform: none; widows: 2; word-spacing: 0px; -webkit-text-stroke-width: 0px; text-decoration-style: initial; text-decoration-color: initial; margin-top: 0px; margin-bottom: 10px; padding: 9.5px; border-radius: 4px; background-color: rgb(245, 245, 245); box-sizing: border-box; overflow: auto; font-size: 13px; line-height: 1.42857; color: rgb(51, 51, 51); word-break: break-all; border: 1px solid rgb(204, 204, 204); font-family: "Times New Roman";">#!/usr/bin/env python3
# -*- coding: utf-8 -*-
#初始传递命令
input_file = 'Bacillus_subtilis.str168.fasta'
list_file = 'list.txt'
output_file = 'gene.fasta'
name_detail = True
##读取文件
#读取基因组序列
seq_file = {}
with open(input_file, 'r') as input_fasta:
for line in input_fasta:
line = line.strip()
if line[0] == '>':
seq_id = line.split()[0]
seq_file[seq_id] = ''
else:
seq_file[seq_id] += line
input_fasta.close()
#读取列表文件
list_dict = {}
with open(list_file, 'r') as list_table:
for line in list_table:
if line.strip():
line = line.strip().split('\t')
list_dict[line[0]] = [line[1], int(line[2]) - 1, int(line[3]), line[4]]
list_table.close()
##截取序列并输出
#定义函数,用于截取反向互补
def rev(seq):
base_trans = {'A':'T', 'C':'G', 'T':'A', 'G':'C', 'N':'N', 'a':'t', 'c':'g', 't':'a', 'g':'c', 'n':'n'}
rev_seq = list(reversed(seq))
rev_seq_list = [base_trans[k] for k in rev_seq]
rev_seq = ''.join(rev_seq_list)
return(rev_seq)
#截取序列并输出
output_fasta = open(output_file, 'w')
for key,value in list_dict.items():
if name_detail:
print('>' + key, '[' + value[0], value[1] + 1, value[2], value[3] + ']', file = output_fasta)
else:
print('>' + key, file = output_fasta)
seq = seq_file['>' + value[0]][value[1]:value[2]]
if value[3] == '+':
print(seq, file = output_fasta)
elif value[3] == '-':
seq = rev(seq)
print(seq, file = output_fasta)
output_fasta.close()</pre>
编辑该脚本后运行,输出新的fasta文件“gene.fasta”,其中的序列即为我们所想要得到的目的基因序列。

扩展:
网盘附件“seq_select.py”为添加了命令传递行的python3脚本,可在shell中直接进行目标文件的I/O处理。该脚本可指定输入fasta序列文件以及记录有所需提取序列位置的列表文件,输出的新fasta文件即为提取出的序列。
#!/usr/bin/env python3
# -*- coding: utf-8 -*-
#导入模块,初始传递命令、变量等
import argparse
parser = argparse.ArgumentParser(description = '\n该脚本用于在基因组特定位置截取序列,需额外输入记录有截取序列信息的列表文件', add_help = False, usage = '\npython3 seq_select.py -i [input.fasta] -o [output.fasta] -l [list]\npython3 seq_select.py --input [input.fasta] --output [output.fasta] --list [list]')
required = parser.add_argument_group('必选项')
optional = parser.add_argument_group('可选项')
required.add_argument('-i', '--input', metavar = '[input.fasta]', help = '输入文件,fasta 格式', required = True)
required.add_argument('-o', '--output', metavar = '[output.fasta]', help = '输出文件,fasta 格式', required = True)
required.add_argument('-l', '--list', metavar = '[list]', help = '记录“新序列名称/序列所在原序列ID/序列起始位置/序列终止位置/正链(+)或负链(-)”的文件,以 tab 作为分隔', required = True)
optional.add_argument('--detail', action = 'store_true', help = '若该参数存在,则在输出 fasta 的每条序列 id 中展示序列在原 fasta 中的位置信息', required = False)
optional.add_argument('-h', '--help', action = 'help', help = '帮助信息')
args = parser.parse_args()
##读取文件
#读取基因组序列
seq_file = {}
with open(args.input, 'r') as input_fasta:
for line in input_fasta:
line = line.strip()
if line[0] == '>':
seq_id = line.split()[0]
seq_file[seq_id] = ''
else:
seq_file[seq_id] += line
input_fasta.close()
#读取列表文件
list_dict = {}
with open(args.list, 'r') as list_file:
for line in list_file:
if line.strip():
line = line.strip().split('\t')
list_dict[line[0]] = [line[1], int(line[2]) - 1, int(line[3]), line[4]]
list_file.close()
##截取序列并输出
#定义函数,用于截取反向互补
def rev(seq):
base_trans = {'A':'T', 'C':'G', 'T':'A', 'G':'C', 'a':'t', 'c':'g', 't':'a', 'g':'c'}
rev_seq = list(reversed(seq))
rev_seq_list = [base_trans[k] for k in rev_seq]
rev_seq = ''.join(rev_seq_list)
return(rev_seq)
#截取序列并输出
output_fasta = open(args.output, 'w')
for key,value in list_dict.items():
if args.detail:
print('>' + key, '[' + value[0], value[1] + 1, value[2], value[3] + ']', file = output_fasta)
else:
print('>' + key, file = output_fasta)
seq = seq_file['>' + value[0]][value[1]:value[2]]
if value[3] == '+':
print(seq, file = output_fasta)
elif value[3] == '-':
seq = rev(seq)
print(seq, file = output_fasta)
output_fasta.close()
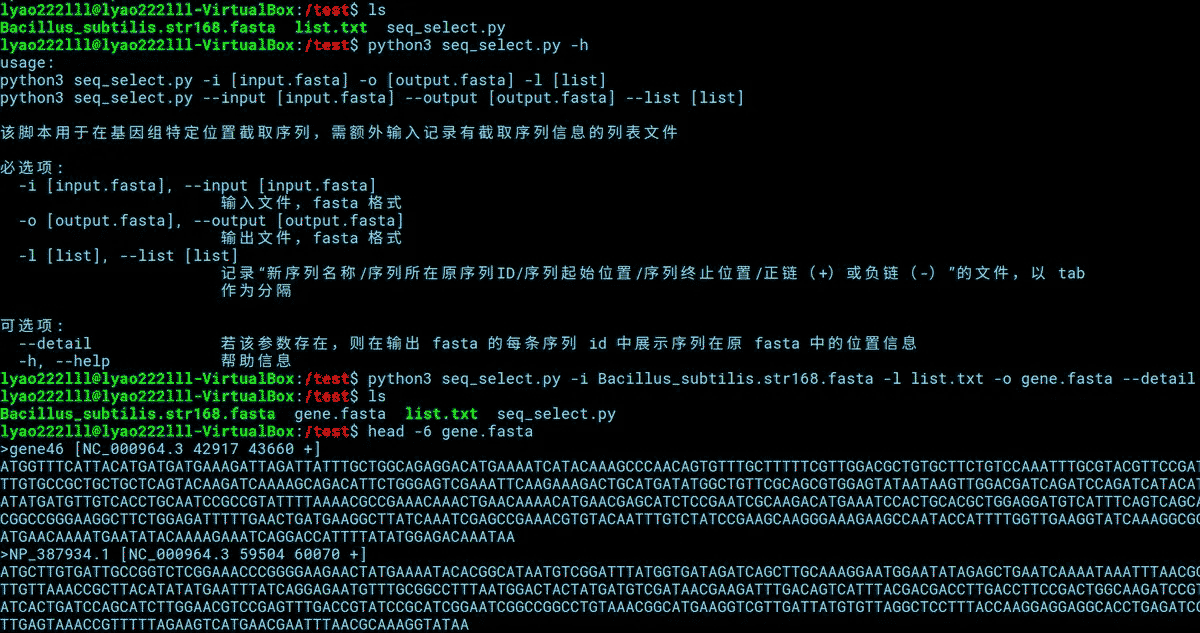
适用上述示例中的测试文件,运行该脚本的方式如下。
#python3 seq_select.py -h python3 seq_select.py -i Bacillus_subtilis.str168.fasta -l list.txt -o gene.fasta --detail
源码提取链接: https://pan.baidu.com/s/1kUhBTmpDonCskwmpNIJPkA?pwd=ih9n
提取码: ih9n
以上就是使用Python脚本提取基因组指定位置序列的详细内容,更多关于python提取基因组位置序列的资料请关注我们其它相关文章!
相关推荐
-
python脚本实现mp4中的音频提取并保存在原目录
一段把mp4中的音频提取为mp3并保存在原目录的python脚本 需要提前安装好ffmpeg 转换为单线程,耗时较长 github: https://github.com/cuifeiran/extract-mp3-from-mp4 #!/usr/bin/env python3 # -*- coding: utf-8 -*- # @Time : 2020/2/26 17:36 # @Author : CuiFeiran # @FileName : tool.py # @Software : Py
-

Python脚本提取fasta文件单序列信息实现
目录 Python脚本编辑 使用的文件 输入 sys模块 从命令行获得文件名称 进行序列信息统计的函数 使用def制作一个函数 .format使用: 进行函数计算 结果屏幕展示 结果输出文件 脚本运行 Python脚本编辑 使用Python对fasta格式的序列进行基本信息统计 预期设计输出文件中包括fasta文件名,序列长度,GC含量以及ATCG各自的含量. 使用的文件 test.fasta stat.py 输入 sys模块 #!/usr/bin/env python import sys 从
-
十个简单使用的Python自动化脚本分享
目录 1.给照片添加水印 2.检测文本文件的相似性 3.对文件内容进行加 密 4.将照片转换为PDF 5.修改照片的长与宽 6.对于照片的其他操作 7.测试网速 8.货币汇率的转换 9.生成二维码 10.制作一个简单的网页应用 在日常的工作学习当中,我们总会遇到各式各样的问题,其中不少的问题都是一遍又一遍简单重复的操作,不妨直接用Python脚本来自动化处理,今天小编就给大家分享十个Python高级脚本,帮助我们减少无谓的时间浪费,提高工作学习中的效率. 1.给照片添加水印 给照片添加水印的代码
-
Python脚本实现定时任务的最佳方法
目录 前言 问题描述 解决方案 总结 前言 在日常工作中,常常需要周期性地执行某些任务,常用的方式是采用 Linux 系统自带的 crond 结合命令行实现,但最近却遇到了一个让人头大的问题. 问题描述 一个包含cx_Oracle的python文件,直接在linux下使用python命令可以运行,但是设置crontab定时任务会报错如下: cx_Oracle.DatabaseError: DPI-1047: 64-bit Oracle Client library cannot be loade
-
使用Python编写提取日志中的中文的脚本的方法
由于工作需要在一大堆日志里面提取相应的一些固定字符,如果单纯靠手工取提取,数据量大,劳心劳力,于是自然而然想到了用Python做一个对应的提取工具,代替手工提取的繁杂,涉及中文字符,正则表达式不好匹配,但不是不可以实现,这个以后优化时再说. 需求描述: 一个父目录中存在多个子文件夹,子文件夹下有多个txt形式化的Log日志,要求从所有地方Log日志中找出CardType=9, CardNo=0时的CardID的值,并将其统计存储到一个文本文件中,要求CardID不能够重复. 需求解析: 首先获取
-
使用Python脚本提取基因组指定位置序列
引言 在基因组分析中,我们经常会有这么一个需求,就是在一个fasta文件中提取一些序列出来.有时这些序列是一段完整的序列,而有时仅仅为原fasta文件中某段序列的一部分.特别是当数据量很多时,使用肉眼去挑选序列会很吃力,那么这时我们就可以通过简单的编程去实现了. 例如此处在网盘附件中给定了某物种的全基因组序列(0-refer/ Bacillus_subtilis.str168.fasta),及其基因组gff注释文件(0-refer/ Bacillus_subtilis.str168.gff).
-
Python 实现字符串中指定位置插入一个字符
如下所示: str_1='wo shi yi zhi da da niu/n'str_list=list(str_1) nPos=str_list.index('/') str_list.insert(nPos,',') str_2="".join(str_list) print(str_2) 从文件中提取行,在行最末尾插入一个逗号. 以上这篇Python 实现字符串中指定位置插入一个字符就是小编分享给大家的全部内容了,希望能给大家一个参考,也希望大家多多支持我们. 您可能感兴趣的文章
-
使用Python向DataFrame中指定位置添加一列或多列的方法
对于这个问题,相信很多人都会很困惑,本篇文章将会给大家介绍一种非常简单的方式向DataFrame中任意指定的位置添加一列. 在此之前或许有不少读者已经了解了最普通的添加一列的方式,如下: import pandas as pd feature = pd.read_csv("C://Users//Machenike//Desktop//xzw//lr_train_data.txt", delimiter="\t", header=None, usecols=[0, 1
-
python读取word 中指定位置的表格及表格数据
1.Word文档如下: 2.代码 # -*- coding: UTF-8 -*- from docx import Document def readSpecTable(filename, specText): document = Document(filename) paragraphs = document.paragraphs allTables = document.tables specText = specText.encode('utf-8').decode('utf-8') f
-
Python实现提取Excel指定关键词的行数据
目录 一.需求描述 1.图片展示 2.提取方法 二.python提取第二版 1.图片展示 2.提取方法 一.需求描述 1.图片展示 从如图所示的数据中提取含有"python"."ubuntu"关键词的所有行数据,其它的不提取: 备注: 关键词和数据行列数可自定义!!! 提取前: 提取后: 2.提取方法 代码如下(示例): import xlrd import xlwt data = xlrd.open_workbook(r'shuju.xlsx') rtable =
-
用Python脚本来删除指定容量以上的文件的教程
文件多了乱放, 突然有一天发现硬盘空间不够了, 于是写了个python脚本搜索所有大于10MB的文件,看看这些大文件有没有重复的副本,如果有,全部列出,以便手工删除 使用方式 加一个指定目录的参数 比如python redundant_remover.py /tmp 主要用到了stat模块,os.sys系统模块 import os, sys #引入统计模块 from stat import * BIG_FILE_THRESHOLD = 10000000L dict1 = {} # filesiz
-
利用python脚本提取Abaqus场输出数据的代码
笔者为科研界最后的摆烂王,目前利用python代码对Abaqus进行二次开发尚在学习中.欢迎各位摆烂的仁人志士们和我一起摆烂!ps:搞什么科研,如果不是被逼无奈,谁要搞科研! 该代码是学习过程中,对前人已有工作所做的稍加修改.为什么是稍加修改,是因为原代码跑不出来!!笔者在提取场输出的位移数据时,渴望偷懒,打算百度一下草草了事,奈何发现网上代码多半驴头不对马嘴,笔者明明是想提取位移,而不是节点和单元的集合!!所以被逼无奈之下,只好硬着头皮修改!欢迎各位大佬们把小弟代码更优化,然后也发给小弟,让小
-
Python在groupby分组后提取指定位置记录方法
在进行数据分析.数据建模时,我们首先要做的就是对数据进行处理,提取我们需要的信息.下面为大家介绍一些groupby的用法,以便能够更加方便地进行数据处理. 我们往往在使用groupby进行信息提取时,往往是求分组后样本的一些统计量(max.min,var等).如果现在我们希望取一下分组后样本的第二条记录,倒数第三条记录,这个该如何操作呢?我们可以通过first.last来提取分组后第一条和最后一条样本.但如果我们要取指定位置的样本,就没有现成的函数.需要我们自己去写了.下面我就为大家介绍如何实现
-
python脚本替换指定行实现步骤
python脚本替换指定行实现步骤 本文主要介绍了Python的脚本替换,由于工作的需要,必须对日志系统进行更新,这里在网上搜索到一篇文章比较不错,这里记录下,大家可以参考下, 工作中需要迁移代码,并把原来的日志系统更新到现在的格式,原来获取log的格式是 AuctionPoolLoggerUtil.getLogger() 现在获取log的格式是: LoggerFactory.getLogger(XXXXX.class) 这里的XXXXX需要替换为当前的类名.如果这样的java文